Phénylcétonurie - signes classiques de transmission héréditaire et de diététique
Sommaire
- 1Comment se manifeste la phénylcétonurie
- 2Mécanisme de développement de la maladie
- 3Phénylcétonurie chez l'enfant
- 4Symptômes de la maladie
- 5Causes et déclencheurs
- 6Diagnostic
- 7Traitement de la phénylcétonurie classique
- 8Caractéristiques de la nutrition du nouveau-né etdiététique
- 9Régime alimentaire pour les enfants d’âge préscolaire et les écoliers
- 10Groupes de produits atteints de PCU
- 11Comment contrôler le taux de phénylalanine dans le sang
- )12Vidéo
Les maladies dont l’apparition est liée à des anomalies de l’appareil cellulaire génétique - la phénylcétonurie - figurent dans une petite liste de maladies héréditaires pouvant être traitées..Le médecin pionnier de cette maladie était un médecin norvégien IA Felling, il a été découvert par la suite qu'un gène unique appelé gène phénylalanine hydroxylase (le bras long du 12ème chromosome contenant jusqu'à 4,5% de tout le matériel ADN cellulaire) était responsable du développement et de l'évolution de la maladie.Un défaut héréditaire entraîne une désactivation partielle ou complète de l'enzyme hépatique phénylalanine-4-hydroxylase.
Comment la maladie de la phénylcétonurie est-elle détectée
La phénylcétonurie héréditaire (PCU) entraîne un empoisonnement chronique du corps par des substances toxiques résultant d'un métabolisme et d'un processus altérés des acides aminéshydroxylation de la phénylalanine.L'intoxication permanente provoque des lésions du système nerveux central (SNC), qui se manifeste par un déclin progressif de l'intelligence (oligophrénie phénylpyruvique).
La maladie de Felling se manifeste par une accumulation excessive de phénylalanine dans l'organisme et par les produits résultant d'un métabolisme incorrect.Le développement de la phénylcétonurie comprend également une altération du transport des acides aminés à travers la barrière hémato-encéphalique, un faible nombre de neurotransmetteurs (sérotonine, histamine, dopamine).En l'absence de traitement rapide de la maladie, il en résulte un retard mental et peut entraîner la mort de l'enfant.

Le mécanisme du développement de la maladie
La cause des troubles géniques est un blocage métabolique qui empêche la formation de phénylalanine-4-hydroxylase (une enzyme,responsable de la conversion de l’acide aminé phénylalanine en tyrosine).La tyrosine, un acide aminé protéinogène, fait partie intégrante des protéines et du pigment mélanique. Elle est donc un élément nécessaire au fonctionnement de tous les systèmes de l'organisme et son absence entraîne une fermentopathie.
L'inhibition de la formation de métabolites causée par l'inactivation mutationnelle de l'enzyme est l'activation des voies métaboliques auxiliaires de la phénylalanine.Un acide aminé alpha aromatique, résultant de processus métaboliques défectueux, se scinde en dérivés toxiques qui, dans des conditions normales, ne forment pas:
- acide phénylpyruvique (phénylpyruvate) - un acide alpha-céto aromatique grasse, sa formationconduit à la myélinisation des processus neuronaux et de la démence;
- L'acide phényl-lactique est un produit formé lors de la récupération de l'acide phénylpyruvique.
- Phényléthylamine - le composé initial pour les émetteurs d’impulsions électrochimiques biologiquement actifs, augmente la concentration de dopamine, d’adrénaline et de noradrénaline;
- L'acétate d'orthophényle est une substance toxique qui provoque des troubles métaboliques dans le cerveau.
Les statistiques médicales indiquent qu'un gène modifié sur le plan pathologique est présent dans 2% de la population, mais il ne se manifeste en aucune manière.L'anomalie génétique est transmise à l'enfant par les parents uniquement en présence de la maladie chez les deux partenaires. L'enfant devient dans 50% des cas le porteur du gène muté et reste en bonne santé.La probabilité que la phénylcétonurie conduise à une maladie chez le nouveau-né est de 25%.
Par quel type est hérité
La maladie de l'abattage est une anomalie génétique héritée d'un type autosomique récessif.Ce type d'héritage signifie que l'apparition des signes d'une maladie congénitale ne se produira que si une copie de gène défectueuse des deux parents, porteurs hétérozygotes du gène modifié, est héritée de l'enfant.
Le développement d'une maladie congénitale dans 99% des cas est dû à une mutation du gène responsable du codage de l'enzyme responsable de la synthèse de la phénylalanine-4-hydroxylase (phénylcétonurie classique).Jusqu'à 1% des maladies génétiques sont liées à des changements mutationnels se produisant dans d'autres gènes qui causentinsuffisance en dihydropteridine réductase (PCU type II) ou en tétrahydrobioptérine (PCU type III).
La phénylcétonurie chez les enfants
La forme classique de maladie génétique chez les enfants se manifeste dans la plupart des cas de façon visible, à partir de 3 à 9 mois.Les nouveau-nés avec un gène défectueux, ont l’air en bonne santé, le trait spécifique est l’apparence spécifique du bébé.La symptomatologie exprimée apparaît 6 à 12 mois après la naissance.
La PCU de type II est caractérisée par le fait que les premiers symptômes cliniques apparaissent un an et demi après la naissance.Les signes de la maladie ne disparaissent pas après le diagnostic d'anomalies génétiques et le début du traitement par un régime.Ce type de maladie congénitale conduit souvent à une issue fatale pendant 2 à 3 ans de la vie d'un enfant.Les symptômes les plus courants de la PCU de type II sont les suivants:
- troubles mentaux graves;
- hyperreflexia;
- altération de la motricité de toutes les extrémités;
- syndrome de contractions musculaires incontrôlées.
Les signes cliniques de modifications mutationnelles des gènes de type III sont similaires à ceux d'une maladie de type II.La carence en tétrahydrobioptérine est caractérisée par une triade de symptômes spécifiques:
- , un degré élevé de retard mental;
- la taille du crâne est nettement réduite par rapport aux autres parties du corps;
- spasticité musculaire (perte totale de mobilité des membres possible).

Manifestations de la maladie de l'abattage
Lors d'essais et d'observations cliniques, il a été suggéré de:Les dérivés toxiques du métabolisme de la phénylalanine entraînent une diminution de la capacité intellectuelle, de nature progressive et pouvant entraîner une démence (oligophrénie, idiotie).Parmi les causes probables de désordres irréversibles de l'activité cérébrale les plus raisonnables, on considère que la diminution du niveau de tyrosine est associée à l'absence de neurotransmetteurs qui transmettent des impulsions entre neurones.
La relation de cause à effet exacte entre maladie héréditaire et troubles cérébraux n'a pas encore été identifiée, de même que le mécanisme de développement dû à la phénylcétonurie d'états mentaux tels que l'échopraxie, l'écholalie, les attaques féroces et l'irritabilité.Les résultats de l'analyse indiquent que la phénylalanine a un effet toxique direct sur le cerveau, ce qui peut également entraîner une diminution de l'intelligence.
Structure et caractéristiques phénotypiques
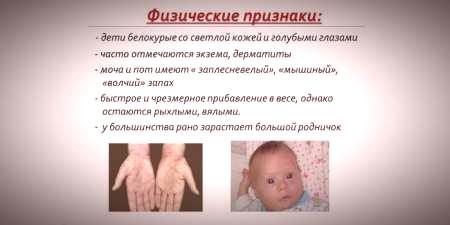
Étant donné que la saturation des pigments dans la peau et les cheveux dépend du taux de tyrosine dans les mitochondries des hépatocytes et que la phénylcétonurie entraîne l'arrêt de la conversion de la phénylalanine, les patients atteints de cette maladie présentent des particularités.signes récessifs).L'augmentation du tonus musculaire provoque l'apparition de déviations dans la structure du corps - elle devient dysplasique.Les caractéristiques externes distinctives de la phénylcétonurie comprennent:
- hypopigmentation - peau claire, yeux bleu pâle, cheveux décolorés;
- cyanose des extrémités;
- réduction de la taille de la tête;
- Position spécifique du corps - lorsqu'il tente de se tenir debout ou assis, l'enfant adopte une posture "sur mesure" (bras et jambes pliés au niveau des articulations).
Symptômesmaladie
Grâce à une détection opportune, la maladie de Felling est traitée avec succès en ajustant la nutrition et le développement de l'enfant se produit en fonction de son groupe d'âge.La difficulté de détecter une mutation génétique réside dans le fait qu'il est difficile de détecter les signes précoces, même par un pédiatre expérimenté.La gravité des symptômes de la maladie congénitale augmente à mesure que l'enfant grandit, car la consommation d'aliments protéinés contribue au développement de troubles du système nerveux central.
Signes chez le nouveau-né
Au cours des premiers jours de la vie d'un enfant, les signes d'anomalies anormales sont difficiles à détecter - le bébé se comporte naturellement, il n'y a pas de retard de développement.Les symptômes de la maladie commencent à apparaître pour la première fois 2 à 6 mois après la naissance.Les parents doivent être attentifs au comportement du bébé, caractérisé par une activité faible, une léthargie ou, au contraire, une anxiété, une hyper-excitabilité.
Dès le début de l'allaitement, les nouveau-nés laiteux commencent à pénétrer dans le corps du nouveau-né avec du lait, ce qui est un catalyseur de l'apparition des premiers signes, ce qui indique clairement que la maladie a commencé à progresser.Les manifestations cliniques spécifiques de la maladie comprennent:
- des vomissements constants (souvent considéré comme un rétrécissement congénital du gardien de but);
- vomissements fréquents;
- aucune réponse aux stimuli externes;
- dystonie musculaire (diminution de la tension musculaire);
- syndrome convulsif (convulsions de nature épileptique ou non épileptique).
Symptômes chez les enfants après 6 mois
Si la maladie génétique ne se manifeste pass'est produite (ou n'a pas été observée) au cours des six premiers mois suivant la naissance de l'enfant, puis, après cette période, il est déjà possible de déterminer avec précision le retard dans le développement psychomoteur.Les symptômes de désordres génétiques causés par un déficit enzymatique chez les enfants de plus de six mois sont les suivants:
- diminution de l'activité (jusqu'à l'indifférence totale);
- manque de maîtrise de soi, d'assise;
- une odeur cutanée particulière de «souris» (l'odeur de moisissure résulte de l'excrétion de dérivés toxiques de la phénylalanine par les glandes sudoripares et l'urine);
- perte de capacité à reconnaître visuellement le visage des parents;
- desquamation de la peau;
- dermatite, eczéma, sclérodermie.
Progression de la maladie en l'absence de traitement pendant l'enfance
Si des anomalies du développement n'étaient pas détectées dans l'enfance et qu'un traitement approprié n'était pas appliqué, alorsla maladie commence à progresser activement et conduit souvent à une invalidité.L'absence de traitement à un stade précoce de la maladie provoque le développement des symptômes suivants à l'âge de 1,5 ans:
- microcéphalie (diminution de la taille du cerveau);
- prognathie (déplacement de la dentition supérieure vers l'avant);
- dentition tardive;
- hypoplasie de l'émail (amincissement ou absence totale d'émail des dents);
- le retard du développement du langage jusqu'à l'absence complète de langage;
- 3, 4 degré d'oligophrénie (retard mental, retard mental);
- cardiopathies congénitales (anomalies de la structure du muscle cardiaque, de parties du cœur, degrands navires);
- troubles du système autonome (acrocyanose, sudation, hypotension artérielle);
- constipation.
Causes et facteurs provocateurs
Pour que se manifeste une mutation autosomique récessive, un gène défectueux doit être hérité des deux parents.Les maladies génétiques de ce type se produisent avec la même fréquence chez les garçons et les filles nouveau-nés.La pathogenèse de la PCU est causée par une altération du métabolisme de la phénylalanine, qui peut se présenter sous 3 formes.La thérapie diététique n’est soumise qu’à la phénylcétonurie classique de type I.
Il est possible de guérir les formes atypiques de la maladie en adaptant la nutrition.Ces déviations sont dues à une carence en tétrahydropterine, déhydropterine réductase (rarement - pyruvyltétrahydropterine synthase, guanosine-5-triphosphate et autres).La plupart des cas mortels sont enregistrés chez des patients présentant des variations rares de la PCU, les manifestations cliniques de toutes les formes de la maladie étant similaires.Le risque d'avoir un bébé avec un gène muté de phénylalanine hydroxylase augmente si les parents sont des proches parents (dans des mariages très proches).
Diagnostic
En cas de suspicion de troubles génétiques, le diagnostic est posé sur la base d'un ensemble de données issues de l'étude des antécédents médicaux - données généalogiques, résultats d'études génétiques cliniques et médicales.Pour détecter rapidement les maladies congénitales (PCU, fibrose kystique, galactosémie, etc.), un programme de masse obligatoire a été mis au pointexamen de laboratoire de tous les nouveau-nés (dépistage néonatal).
Si les futurs parents sont informés du porteur du gène muté, la médecine moderne offre des moyens de détecter un défaut au stade de la grossesse (diagnostic prénatal du fœtus par une méthode invasive).Pour classer les types de phénylcétonurie en fonction de leur gravité, une classification conditionnelle basée sur le taux de phénylalanine dans un liquide plasmatique sanguin exempt de fibrinogène est utilisée:
- Phénylcétonurie lourde - 1200 μmol /l.
- Moyenne - 60-1200 µmol /l.
- Léger (aucun traitement requis) - 480 μmol /L.
Test de dépistage
La détection des anomalies génétiques s'effectue en plusieurs étapes.À la première étape de la maternité, tous les nourrissons âgés de 3 à 5 jours sont soumis à un prélèvement de sang périphérique (sur cinq) à des fins de recherche.Le matériau est appliqué sur un support papier et envoyé au laboratoire biochimique où il fait l'objet d'une analyse biochimique.Dans la deuxième étape du test de dépistage, la concentration de concentration normale en phénylalanine est déterminée.

Si aucun changement pathologique n'est détecté, le diagnostic est établi et la carte de l'enfant est enregistrée.En cas d'écarts par rapport à la norme, les résultats du diagnostic sont envoyés au pédiatre pour assurer un examen plus précis de l'échantillon de sang du nouveau-né.La santé de l'enfant dépend de la mise en œuvre rapide et précise de toutes les mesures permettant de détecter les écarts.Si le diagnostic est confirmé après des dépistages répétés, les parents de l'enfantenvoyé à une clinique de génétique pédiatrique pour traitement.
Analyses et études permettant de confirmer le diagnostic.
Un nouveau diagnostic lors de la détection lors du test de dépistage principal des anomalies est effectué en soumettant à nouveau les tests.Outre la détermination de la teneur en phénylalanine dans le sang pour les méthodes de diagnostic de la PCU chez les enfants et les adultes, citons:
- Test de corroyage - détermination de l’acide phénylpyruvique dans les urines en ajoutant du chlorure de fer au biomatériau (coloration en couleur bleu-vert);
- Test de Guthrie - Évaluation du degré de réponse des micro-organismes au métabolisme ou aux enzymes contenus dans le sang du patient;
- chromatographie - étude des propriétés chimiques de substances réparties entre deux phases;
- fluorimétrie - irradiation de biomatériau par un rayonnement monochromatique afin de déterminer la concentration de substances qui y sont contenues;
- électroencéphalographie - diagnostic de l'activité électrique du cerveau;
- L'imagerie par résonance magnétique est l'excitation des noyaux atomiques de cellules par des ondes électromagnétiques et la mesure de leur réponse.
Traitement de la phénylcétonurie classique
Au cœur du traitement par la phénylcétonurie, il existe une restriction à la consommation de produits à la source de protéines animales et végétales.La seule méthode de traitement efficace est la thérapie par le régime dont l’adéquation est évaluée par la teneur en phénylalanine dans le sérum.Niveau maximal d'acides aminés chez les patients de différents groupes d'âgeest:
- chez les nourrissons et les enfants jusqu'à 3 ans - jusqu'à 242 µmol /l;
- chez les enfants d’âge préscolaire jusqu’à 360 µmol /l;
- chez les patients âgés de 7 à 14 ans - jusqu'à 480 μmol /l;
- chez les adolescents - jusqu'à 600 μmol /l.
L'efficacité du régime alimentaire dépend du stade de la maladie pour lequel le régime est corrigé.En cas de diagnostic précoce de pathologie congénitale, une diète est prescrite à partir de la 8e semaine de vie (après cette période, des changements irréversibles ont déjà commencé).L'absence de mesures opportunes entraîne des complications et une diminution du niveau d'intelligence de 4 points pendant un mois, de la naissance au début du traitement.

Etant donné que le régime thérapeutique de la phénylcétonurie prévoit une exclusion complète du régime des protéines animales, il est nécessaire d'utiliser d'autres sources d'acides aminés essentiels, ainsi que des vitamines du groupe B, le calcium -et des composés minéraux contenant du phosphore.Les produits à utiliser comme suppléments sans protéines comprennent:
- des hydrolysats de protéines (Amigen, Aminazole, Fibrinosol);
- ne contiennent pas de mélanges de phénylalanine saturés en acides aminés essentiels - Tetrafen, sans phényle.
Outre les mesures curatives visant à éliminer la cause d'altération du fonctionnement du corps, un traitement symptomatique devrait être mis en œuvre dans le but d'éliminer les troubles de la parole et de normaliser la coordination des mouvements.La thérapie complexe comprend les procédures de physiothérapie, les massages, l’aide d’un orthophoniste, d’un psychologue et l’exercice de gymnastique.Dans certains cas, en conjonction avec une thérapie de régimemontre l'utilisation d'anticonvulsivants, de médicaments nootropes et vasculaires.
Particularités du traitement des formes atypiques
Les phénylcétonuries de types II et III ne peuvent pas être traitées avec un régime pauvre en protéines: le taux de phénylalanine dans le sang reste inchangé, le flux de protéines dans le corps reste inchangé ou les symptômes cliniques progressent même avec la diminution du niveau d'acides aminés.Un traitement efficace de ces formes de la maladie est mis en oeuvre avec:
- le facteur tétrahydrobioptérine - facteur de l’enzyme affectée;
- analogues synthétiques de la tétrahydrobioptérine - ces substances pénètrent mieux dans la barrière hémato-encéphalique;
- médicaments de traitement de substitution - n'éliminent pas la cause de la phénylcétonurie, mais favorisent le fonctionnement normal du corps (lévodopa avec Carbidofa, 5-oxytryptophane, 5-formyltétrahydrofolate);
- hépatoprotecteurs - soutiennent la fonction hépatique;
- anticonvulsivants;
- , l'introduction du gène de la phénylalanine hydroxylase dans le foie est une méthode expérimentale.
Nutrition et thérapie diététique du nouveau-né
L'allaitement au sein est autorisé au cours de la première année de vie d'un bébé atteint de PCU, mais doit être restreint.Jusqu'à 6 mois, l'apport acceptable de phénylalanine est compris entre 60 et 90 mg par kg de poids de bébé (100 g de lait contiennent 5,6 mg de phénylalanine).À partir de 3 mois, le régime alimentaire du bébé devrait être progressivement élargi, en y introduisant jus et fruits en purée.
Les enfants à partir de 6 mois sont autorisés à manger des purées de légumes, du porridge (sagou), sans protéines.acide.Après 7 mois, il est possible de donner aux bébés des pâtes faibles en protéines à partir de 8 mois - du pain qui ne contient pas de protéines.L'âge auquel la protéine dans le corps de l'enfant malade devrait être restreinte n'a pas été établi.Les médecins discutent encore de la faisabilité d'une thérapie par le régime pour toute la vie, mais ils conviennent qu'un régime d'au moins 18 ans devrait être suivi.
La phénylcétonurie diagnostiquée chez une femme n'est pas une raison pour refuser la naissance d'un enfant.Femmes enceintes atteintes de PCU afin de prévenir les lésions fœtales pendant la grossesse et d’éventuelles complications, il est nécessaire de suivre un régime avec une alimentation restreinte à la phénylalanine (au moment de l’accouchement) (jusqu’à 242 μmol /l).
Mélanges pour bébés sans lactose
Le régime alimentaire de la phénylcétonurie repose sur une réduction significative de la quantité de protéines naturelles dans l'alimentation quotidienne, mais le corps du nourrisson ne peut pas se développer normalement en l'absence des oligo-éléments nécessaires.Pour répondre aux besoins du bébé en protéines, on utilise des mélanges d’acides aminés sans lactose qui, en vertu de la législation russe, doivent être fournis gratuitement.
La tolérance du nourrisson à la phénylalanine évoluant rapidement au cours de la première année de vie, sa concentration dans le sang du bébé doit être surveillée et le régime ajusté.Les mélanges sont conçus pour certains groupes d'âge:
- sont prescrits aux bébés de moins d'un an: Afenilak 15, Analog-SP, PKU-1, PCU-mix, PCU Anamix;
- Enfants de plus d'un an.année, nommer enrichi avec des mélanges de vitamines et de minéraux à haute teneur en protéines - Prima PCU, P-AM Universal, PKU-1, PKU-2, Maximeid XP, Maximum XP.

Aliments à base de protéines
L’un des principaux composants d’un régime alimentaire contre la phénylcétonurie est constitué de produits à base d’amidon à faible teneur en protéines.Ces suppléments contiennent de l'hydrolysat de caséine, du tryptophane, de la tyrosine, de la méthionine, de l'azote et apportent au bébé un besoin quotidien en protéines nécessaires à son développement et à sa croissance normaux.Les aliments spécialisés qui comblent le manque de minéraux essentiels et d'acides aminés lorsqu'ils manquent de régime sont:
- , Berlofen;
- Zimorgan;
- Minafen;
- Aponti.
Régime alimentaire pour les enfants d’âge préscolaire et les écoliers
À mesure que le corps s’ajuste à la phénylalanine, les enfants à partir de 5 ans peuvent progressivement réduire leurs restrictions alimentaires.L'expansion d'un régime se produit par l'introduction de céréales, produits laitiers, produits à base de viande.Les étudiants du troisième âge ont déjà une grande tolérance à la phénylalanine. Vous pouvez donc, à cet âge, continuer à élargir le régime, tout en surveillant la réaction à toute modification de la nutrition.Les méthodes suivantes sont utilisées pour contrôler l'état de l'enfant:
- , évaluation de paramètres neurologiques, état psychologique;
- contrôle des performances de l'électroencéphalogramme;
- détermination du taux de phénylalanine.
Groupes d'aliments contenant de la PCU
Le régime alimentaire des patients atteints de PCU, associé à une faible teneur en protéines.Les produits amylacés et les mélanges médicinaux incluent également les produits d'origine naturelle.Lors de l'établissement du menu, la quantité de protéines consommée doit être clairement calculée et la posologie recommandée par le médecin ne doit pas être dépassée.Pour éliminer les effets toxiques sur le corps, 3 listes de produits contenant des articles interdits (rouge), non recommandés (orange) et autorisés (vert) ont été développées.
Liste rouge
La phénylcétonurie se développe sur l’absence d’une enzyme qui se transforme en tyrosine phénylalanine. C’est pourquoi une forte teneur en protéines est à la base de l’énumération des produits figurant sur la liste des produits interdits (liste rouge).Les éléments de cette liste devraient exclure totalement le régime alimentaire d'un patient atteint de PCU:
- ;
- organes internes des animaux, sous-produits;
- saucisses, saucisses;
- fruits de mer (y compris le poisson);
- œufs de tous les oiseaux;
- produits laitiers;
- noix;
- légumineuses et céréales;
- produits à base de soja;
- plats à base de gélatine;
- confiseries;
- aspartame.
Liste orange
Les produits à administrer à un enfant chez qui un diagnostic de PCU a été diagnostiqué sont inclus dans la liste orange.L'inclusion dans le régime des éléments de cette liste est acceptable, mais dans un nombre strictement limité.Bien que ces produits ne contiennent pas beaucoup de protéines, ils peuvent également augmenter le taux de phénylalanine. Par conséquent, leur utilisation n'est pas recommandée:
- de légumes en conserve;
- plats à base de pommes de terre et de riz;
- chou;
- lait;
- sorbet.
Liste verte
Les produits sans protéines peuvent être utilisés chez les patients présentant un diagnostic de phénylcétonurie sans restriction.Avant d'acheter les articles figurant sur la liste verte, il est nécessaire d'examiner la composition indiquée sur l'emballage et de s'assurer qu'il n'y a pas de colorant pour l'aspartame contenant de la phénylalanine:
- des fruits;
- légumes (à l'exception des pommes de terre et des choux);
- baies;
- verts;
- céréales amylacées (sagou);
- miel, sucre, confiture;
- produits à base de farine à base de farine de maïs ou de riz;
- huiles, graisses (beurre, tournesol, olive).
Comment contrôler le taux de phénylalanine dans le sang
La phénylcétonurie est une maladie incurable qui peut être transmise à la phase de stagnation par le biais d'un régime alimentaire et de mesures thérapeutiques.Au changement des conditions de vie, la perturbation de l'alimentation peut aggraver la maladie, les patients doivent donc être surveillés toute leur vie durant.Le processus de contrôle consiste à déterminer périodiquement le taux de phénylalanine dans le sang.La fréquence du test dépend de l'âge du patient:
- jusqu'à 3 mois - un test de dépistage sanguin doit être effectué chaque semaine pour obtenir des résultats cohérents.
- de 3 mois à 1 an - 1 à 2 fois par mois;
- 1 à 3 ans - une fois tous les 2 mois;
- plus de 3 ans - trimestriel.

Le sang à analyser est administré 3 à 4 heures après l'ingestion.En plus du dépistage, le développement de la PCU est contrôlé en déterminant l’état nutritionnel, le développement physique et émotionnel du patient, le niveau de ses capacités intellectuelles.et développement du langage.Les observations peuvent nécessiter des diagnostics supplémentaires avec la participation de spécialistes appropriés.
Vidéo
Les informations présentées dans cet article sont à titre indicatif.L'article n'appelle pas à l'auto-traitement.Seul un médecin qualifié peut diagnostiquer et recommander un traitement en fonction des caractéristiques individuelles du patient.












